Metformin is many longevity researchers favorite longevity drug. Harvard professor David Sinclair wrote an entire chapter in his book on longevity about metformin, titled “This cheap pill might help you live a longer, healthier life”. So, it’s probably worth exploring. Let’s get into it.
If you’re a healthy person, should you be taking metformin for longevity?
No, probably not.
Is this because metformin just needs to be developed more, and, in 30 years, we’ll all be taking metformin for longevity?
Also no.
Well, that’s it for today’s blog post. Tune in next week for more.

Of course, if you’d like a bit more information, I can explain my reasoning.
It’s difficult to evaluate metformin based on mechanisms because nobody really knows how it works. We know its results: it decreases glucose production by the liver, increases the insulin sensitivity of body tissues, and reduces appetite. However, we don’t know how it accomplishes any of these results, which means that it’s difficult to say whether it’s effective for anything beyond the obvious (i.e. reducing glucose, increasing insulin sensitivity, and reducing appetite).
There are two ways to look at a drug that has unclear mechanisms like metformin. The first way is to assume that it doesn’t have any effects beyond what’s obvious. The second way is to assume that there are untold mysteries within the mechanisms of that drug, and exhaustively check the side effects and case reports of patients who took the drug to find any possible other beneficial effects of the drug. Substantiated beneficial effects raise the possibility of repurposing the drug for this alternative beneficial effect.
The former path is safer scientifically; the latter path is more fun and carries the possibility of profit. As a huge proponent of drug repurposing myself (it’s what I’ve based my company on), I too would like to have fun and profit. Also, if nobody ever pursued drugs with unclear mechanisms, we’d only have about half of the drugs that help us in our daily lives.
But, I’m too much of a worrier to go blindly off in the pursuit of a good time without some ground rules. The repurposing field is littered with the corpses of research papers of poor scientists who ventured off without ground rules, and got stuck arguing for years that curcumin (the main compound in turmeric) is a cure-all, only to find out that curcumin is just a molecule that interferes a lot with assays.

So, here are my ground rules for being safe while looking for possible repurposing of drugs like metformin:
1. Any proposed repurposed indication needs to be supported by at least a vaguely plausible mechanism AND a human case study OR a retrospective study in humans
2. If a meta-analysis suggests your repurposed indication is wrong, you better have a really, really good reason as to why the meta-analysis is looking at the wrong thing
3. If you want to support your mechanism with animal studies, you better have spent a ton of effort making sure that the animal studies can translate to humans
The reason why I have these rules is to prioritize the highest quality evidence, absent the sort of randomized controlled trial that’s the real gold standard of drug discovery. So, the highest quality evidence is actual, visible, incontrovertible evidence in humans, supported by theory. If the theory gets overturned, that’s a bad sign but not necessarily a dealbreaker. If the evidence in humans gets overturned, that’s generally a really bad sign.
Unfortunately, metformin as a longevity drug fails all these rules. Let me use this review paper as a guide.
First, if we take a look at the proposed mechanism of metformin as a longevity drug, we get a lot of suggestive stuff about metformin impacting “insulin growth factor 1” and “AMP-activated kinase”, leading to extended lifespan. Unfortunately, all of these impacts happen in C. elegans (a.k.a. roundworms). I would have to see a lot of evidence to extend a result from roundworms to humans, but metformin proponents expect me to just take it at face value.
Second, the retrospective studies that try to link metformin to longevity are, generally speaking, not great. The authors try to link the mild weight loss associated with metformin to caloric restriction, then try to argue the long lifespan of Okinawans is due to caloric restriction. Not only is that explanation of the long lifespan of Okinawans arguable, but literally any weight loss drug could be linked to longevity in that way (like, for example, meth).

Finally, and most damningly, the meta-analysis really doesn’t support the indication. The largest meta-analysis of patients with type 2 diabetes on metformin showed basically zero effect of metformin on mortality, cardiovascular disease, or cardiovascular conditions. Given that type 2 diabetics should be the patients who’d benefit most from metformin, this is especially bad.
So, overall, my cautious self would not be overall too eager to follow metformin down the longevity path. If I were cynical, I might write a book or make a podcast advocating it to other people (ahem), but I would not stake my own money on metformin as a longevity drug.
I assume if I showed this evidence to metformin advocates, however, they would not agree that this is an indication that metformin is not a longevity-promoting drug. They might argue that metformin’s impacts on longevity are real, but only exist in the super high doses that we can give mice but not humans. They’d continue to say that metformin’s true capabilities have yet to be unlocked because it’s not fully developed for longevity. Give metformin 30 years of development, they’d say. It’ll get developed into super-metformin just in time for you to avoid needing Medicare.
As you can probably guess, I don’t agree with this. Even if I think there are some latent abilities of metformin to promote longevity (which I doubt), I don’t think it’s likely that we’ll have super-metformin in 30 years, even with the multimillion dollar startups that are no doubt working on it right now. This is because metformin, in its current state, does not fit well onto the pharma development pathway, and no amount of money will change that.
In order to explain this, let me explain how pharma development normally works. Not only will this be informative in terms of what the development of metformin should be (but isn’t), but it’ll also be an interesting exploration of one of the few high-tech fields that actively shares their work with the public.
Instead of talking in the abstract, however, I’ll be using the examples of Lipitor (atorvastatin), the best selling drug of all time, and the recent, super exciting weight loss drug Wegovy (semaglutide). I’ll talk about how they were developed, and then loop back to how metformin can’t be developed in the same way, no matter how many years or dollars we give it.
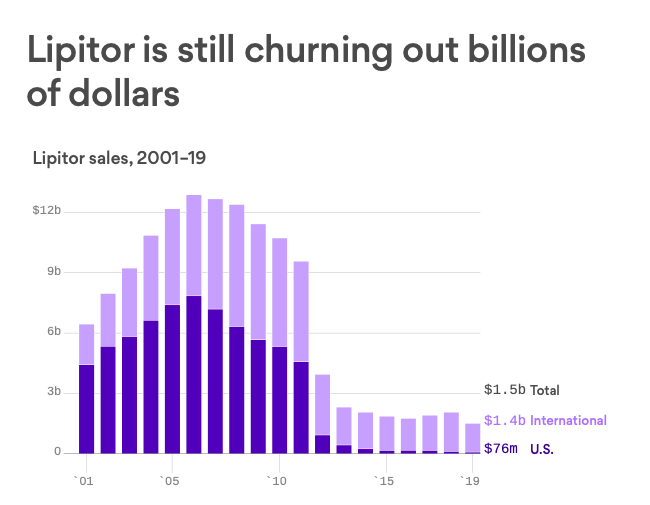
Let’s first start with Lipitor. Fortunately for us, there’s an excellent paper written by Bruce Roth, who was largely responsible for the invention of Lipitor, on how he did it. So, I’ll rely on that.
The invention of Lipitor started with a highly specific understanding of a specific pathway in the body: the cholesterol biosynthesis pathway. From the very beginning of the invention of Lipitor, scientists were aware of the exact pathway, the steps involved in the pathway, and which specific step in the pathway needed to be inhibited in order to safely lower cholesterol in animals or humans.
Starting off with this knowledge had a number of advantages.
First, it allowed them to be able to quickly identify small molecules that did what they needed to do, including the random fungal extract that happened to inhibit exactly the right step in the cholesterol biosynthesis pathway. All subsequent inhibitors were based on that fungal extract.
Second, it gave them confidence on what exact in vitro and animal models they could test any inhibitors on. They tested all variations on that fungal inhibitor on rat, dog, monkey, and human liver while it was being developed. Then, once they picked an ideal inhibitor, they also tested it in live rabbits, dogs, and guinea pigs.
Third, it let the scientists know exactly what to optimize for. They needed an inhibitor that was non-toxic and was as effective as possible on inhibiting that specific pathway. These were the two parameters they optimized for.
With these three advantages, the invention of atorvastatin was a chemistry problem, rather than a biology problem. If you read the account of the inventor, the chemistry was very clever (if you’d like a rundown on this clever chemistry, follow the footnote [1]), but, still, the exact knowledge of the pathway involved made this process solvable with enough work.
The story of the invention of semaglutide follows the same basic outlines, even though semaglutide is a more complex drug that relied on more complex discovery processes. Once again, I’ll be relying on an account of the invention by the inventors themselves.
Semaglutide, unlike atorvastatin, is a peptide hormone, or a short chain of amino acids that is distributed throughout the body. Atorvastatin is just classified as a statin, which is a type of small molecule. Peptides are not only trickier to modify chemically, they’re trickier to even see if you have modified them chemically, as x-ray crystallography doesn’t work with them. To get an idea of why this might be, I’ve put pictures of the chemical structures of atorvastatin and semaglutide below.
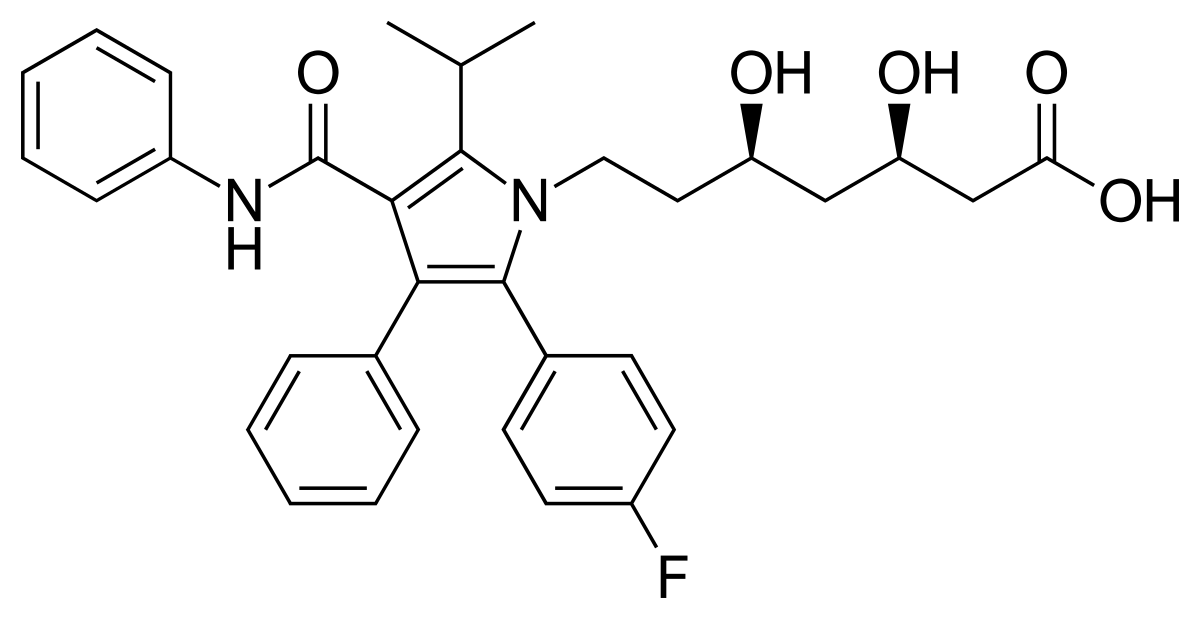
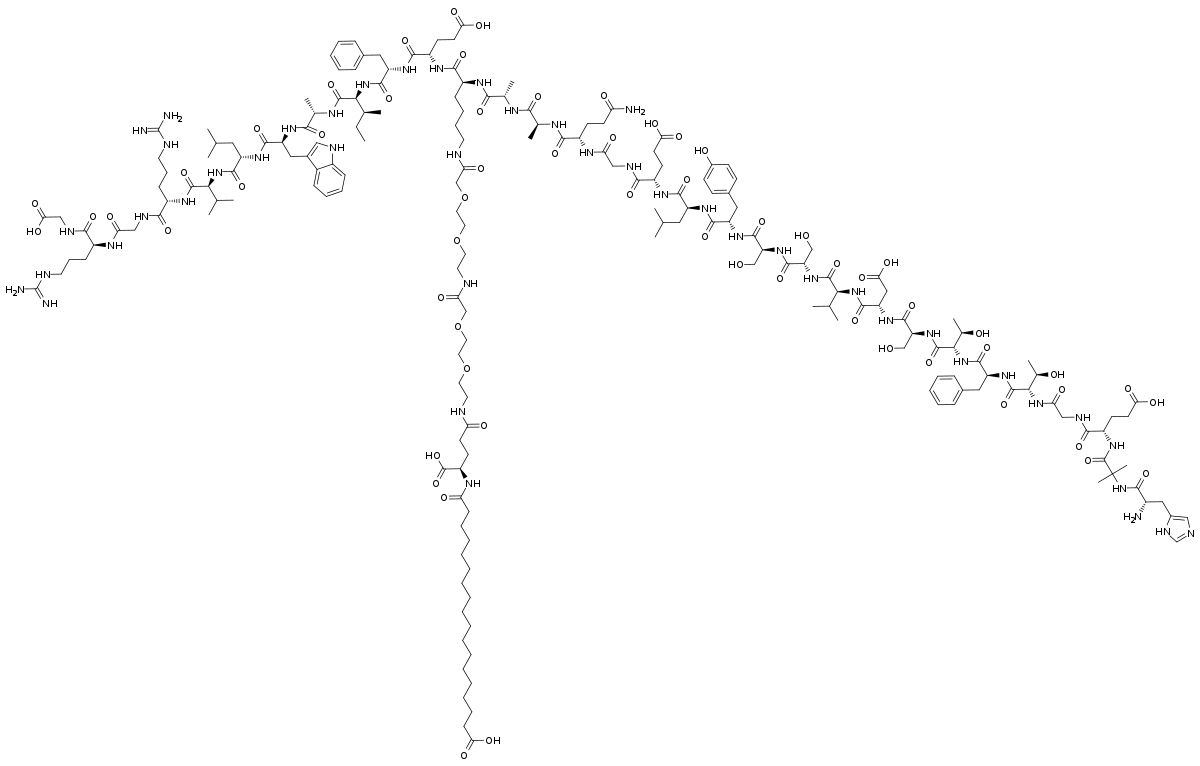
This is why, technically, the development of semaglutide was a much more difficult task than the development of atorvastatin. A number of techniques were used that weren’t available to the atorvastatin team: alanine scanning to be able to determine the structure, attaching fatty acids to facilitate the binding of the peptide to albumin, and cloned human GLP-1 receptors in baby hamster kidney cells to check the binding (for more information on these clever techniques, check out footnote [2]).
However, what’s important to note is what stayed the same between atorvastatin and semaglutide: a deep knowledge of GLP-1, the GLP-1 receptor, and what exactly the function of GLP-1 is in the body. In the case of GLP-1, it was primarily that GLP-1 triggered insulin secretion, and then later that GLP-1 could result in weight loss.
Once again, as they tested their various synthetic versions of GLP-1 on human GLP-1 receptors, rats, minipigs, and mice, they could check for receptor binding, toxicology, and insulin response. There were missteps along the way, like taspoglutide, which made it all the way to phase III trials before they realized it could cause anaphylactic shock. However, they always knew what their north star was.
This is not the case for metformin. Metformin not only lacks a target for longevity, it lacks a target for its main indication, too, type 2 diabetes. Without a clear target, there’s no optimizing, which means there’s no way to put metformin on the pharma development treadmill.
I’m hopeful that fundamental longevity research will start identifying definite pathways and targets for metformin, which would make a huge difference for this drug development process. The only alternative is to rely on some completely different drug development process that allows optimizing for essentially unknown pathways. While AI/machine learning drug development companies promise that this can be done, I will believe it when I see it.
[1] Ok, so here’s the chemistry as he writes it/I understand it.
They knew they needed an inhibitor of cholesterol biosynthesis. However, they were gun-shy because the first attempt at an inhibitor of cholesterol biosynthesis was triparanol, which was introduced in 1959 before extensive toxicology was required for new drugs. Unfortunately, what toxicology would have revealed was that triparanol, by inhibiting the last step in cholesterol biosynthesis, causes a build-up in the penultimate product in the pathway, desmosterol. Accumulation of desmosterol is super bad because there’s no biological way to get rid of it, and eventually you get cataracts and other bad side effects. This ended up being discovered 3 years after triparanol was released to the public and caused a bunch of people to go blind.
So, instead, the hypothesis was that you could inhibit cholesterol earlier in the pathway. Specifically, if you stop the pathway at the point that HMG-CoA turns into mevalonic acid, you end up with a lot of HMG-CoA instead. This is a lot easier to get rid of because HMG-CoA is also part of the ketogenesis pathway, so the body has ways of getting rid of it already.
Everyone was looking for an inhibitor of the specific enzyme that turns HMG-CoA into mevalonic acid, HMGR. A scientist in Japan, Akira Endo, hypothesized that fungi used chemicals to ward off parasitic organisms by inhibiting cholesterol synthesis, based on the fact that fungi use ergosterol instead of cholesterol (so they could inhibit cholesterol synthesis without hurting themselves). Then he studied 6000 compounds isolated from fungi and found 3 that worked, which is one way of doing it.
The most potent chemical that Endo found was called compactin. After he published his findings, everyone else also started looking at fungal metabolites, and soon enough Merck and Sankyo isolated their own fungal metabolites to inhibit cholesterol biosynthesis. Merck also found something interesting: that “ortho-biphenyl containing 3,5-dihydroxy-6-heptenoic acids and their lactones … were equipotent to the fungal metabolites at inhibiting HMGR in vitro.”
Merck publicized this finding in a patent, and probably expected this to be a neat finding and a good way to shore up their own fungal metabolite’s intellectual property (by preventing anyone else from chemically synthesizing something similar). However, Bruce Roth and his team read this, and realized that Merck had just given away the key: this specific part of the fungal metabolite was the only important part to inhibit cholesterol biosynthesis. Everything else was useless.
Or, in Bruce’s own words: “the key requirements for potent inhibition of HMGR were a mevalonolactone/3,5-dihydroxy-heptanoic or -6-heptenoic acid moiety and a large lipophilic group held in the correct spatial relationship by a spacer or template group…virtually any ring system which fulfilled this requirement would lead to a series of potent inhibitors.”
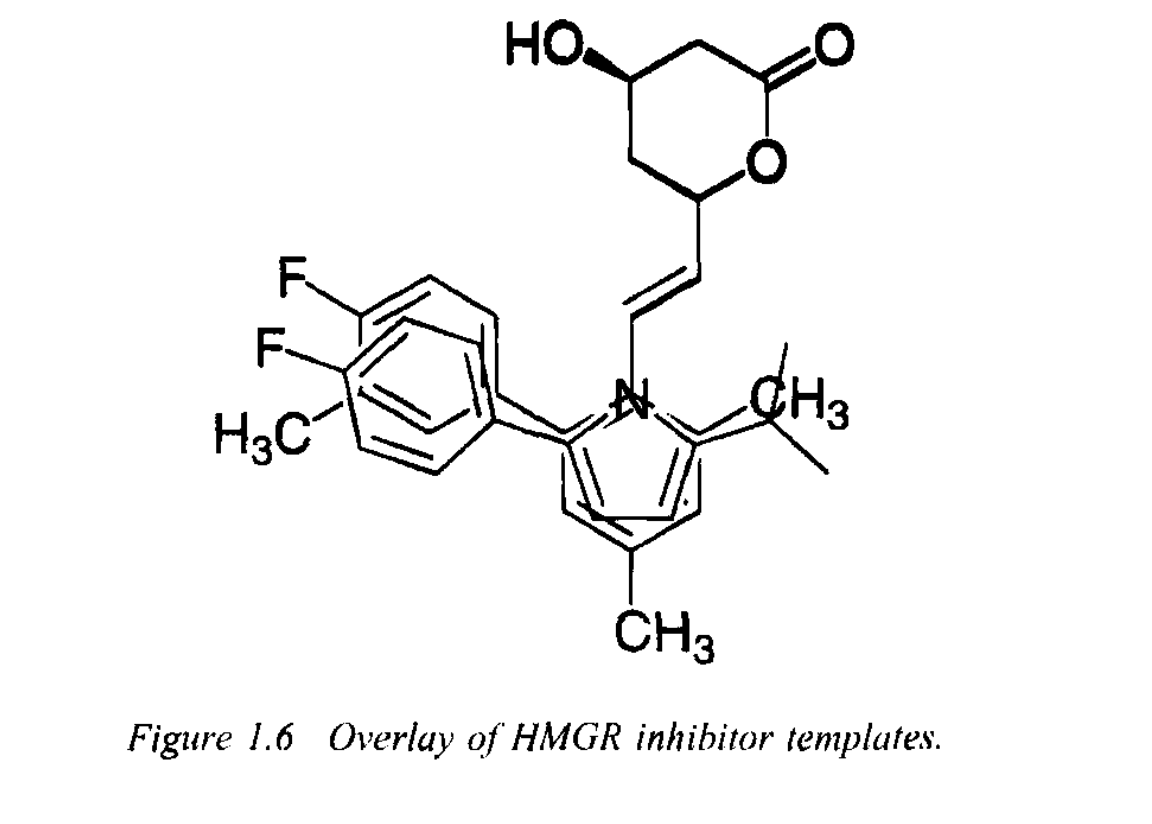
This was the starting point. Then they actually had to figure out the easiest way to construct this ring system. They eventually settled on the 1H-pyrrole system, which they were very familiar with and comfortable with working in. They constructed 30 different versions of this ring to try to figure out the correct spatial relationship. Unfortunately, once they did so and found the best possible version of the ring, they realized that it still wasn’t good enough. It was only 1/10th as good as the fungal metabolite. They (and Merck) must have been missing something else. But what?
They decided to go the simple way about it and just overlay their chemical diagram on top of Merck’s and see what was missing, as seen below. When they did so, they realized they were missing Merck’s methyl group. So, they tried putting in a methyl group, and voila! It was a successful inhibitor!
Of course, in biochemistry, nothing’s ever so easy. As it turned out, this new compound was more effective at inhibition, but it was also way more toxic than the old compound. Instead of giving up on this tact, however, they decided to once again create a bunch of variations on this compound in order to find one that was less toxic. In this case, they thought that the toxicity came from the bromines that had been introduced with the methyl group, so they created 20 different versions with substitutions for the bromines and tested them all in rabbits and dogs. Eventually, they found one: atorvastatin.
They then faced a critical decision: whether to develop the compound as a racemate (a mixture of bioactive and non-bioactive versions of a drug) or the stereoisomer (the pure version). A racemate would be cheaper and easier to develop and manufacture, but the purified stereoisomer would place less stress on the body and require less drug to be effective.
Atorvastatin was already likely to be coming onto the market 10 years after the fungal metabolites by the time they made this decision. One way to look at that fact was, “We need to get this out of the door ASAP before we spend any more money on it.” Bruce took the other point of view, “We need something that’s incredibly good in order to displace the fungal metabolites”.
So, they decided to go the expensive route, and developed the compound as a stereoisomer. Then they tested it in healthy volunteers, and found that it lowered cholesterol more effectively than any other drug on the market. The rest was history.
[2] This stuff is more advanced than the atorvastatin stuff, so I am largely out of my depth. That’s never stopped me before, though, so I’m going to do my best.
The central problem with using biologic GLP-1 as a therapy, as mentioned, was that it had a short half-life. So all efforts were focused on creating a version of GLP-1 that had a long half-life.
The old methods of creating a drug with a long half-life don’t work for a giant peptide like GLP-1. It’s way too big. So, a new way of creating a drug with a long half-life is to bind it to albumin, which is an all-purpose carrier protein in the body. Makes sense, right? Just use the body’s method of carrying proteins and preventing them from breaking down.
In order to get albumin to carry a peptide, though, you need a peptide-based ligand, or something to chemically attach the protein to the albumin. Before any of the stuff with semaglutide, a lot of work had been done to try to find these ligands. The original team built off this work to find ligands that could reversibly bind to albumin (i.e. GLP-1 could be attached to albumin for transport, and then unattached for use).
In order to see what they could attach to GLP-1 without messing up its function, they employed an alanine scan. In an alanine scan, each amino acid in a peptide is subsequently replaced by alanine to see which amino acid actually does. It’s sort of like figuring out how a radio works by taking one piece out at a time then seeing how the radio functions without it.
From there, they found the parts of GLP-1 that were necessary for its function and shouldn’t be messed with, and then the parts that weren’t necessary and could have ligands attached to it. Then, they again used the technique of creating a bunch of versions of GLP-1 with ligands attached to it to see which had both a good attachment to albumin and were still functional.
From this exploration, they created liraglutide, the first effective GLP-1 agonist. Novo Nordisk took this work and improved on it by noticing that compounds that bound really well to albumin bound less well to GLP-1 receptors, probably because there was competition between them. They did stuff I don’t entirely understand with fatty acids to split the difference and come up with semaglutide, which they then tested on diabetic mice. It worked well on mice and had a long half-life, so they knew they had a hit product.