So, the situation with GLP-1 receptor agonism and GIP receptor agonism is weirder than I thought.
To recap, GLP-1 receptor agonists, like Ozempic, cause people to lose weight. GLP-1 receptor/GIP dual receptor agonists, like Mounjaro, cause people to lose even more weight. As such, these dual agonists are often sold as sequels or improvements to mono agonists.
But, as I wrote previously, the story is more complicated than just “dual agonism is better than single”, because GLP-1 receptor agonism combined with GIP receptor inhibition is also more effective at causing weight loss than GLP-1 receptor agonism alone. This has been shown in mice and now Amgen has claimed they have shown it in humans (and have also claimed that the weight loss with this agonist/inhibitor drug is more sustained than the weight loss of agonist/agonist drug, but that’s a separate issue I will cover in a later blog post).
Thanks for reading Trevor Klee’s Newsletter! Subscribe for free to receive new posts, which tend to be about obesity a surprising amount. What can I say: it’s an interesting subject.
Subscribed
Well, as it turns out, the story is even more complicated than that, at least according to this great review paper. First of all, apparently, it works in the reverse: GLP-1 receptor inhibition can result in weight loss (in mice), so, that’s pretty weird. Second, remember how GLP-1 receptor agonism results in insulin secretion and weight loss? Well, apparently, those two don’t always have to be correlated, when you do different combinations of agonism and inhibition.
Confused, yet? Because I am. I was so confused that I made a table.
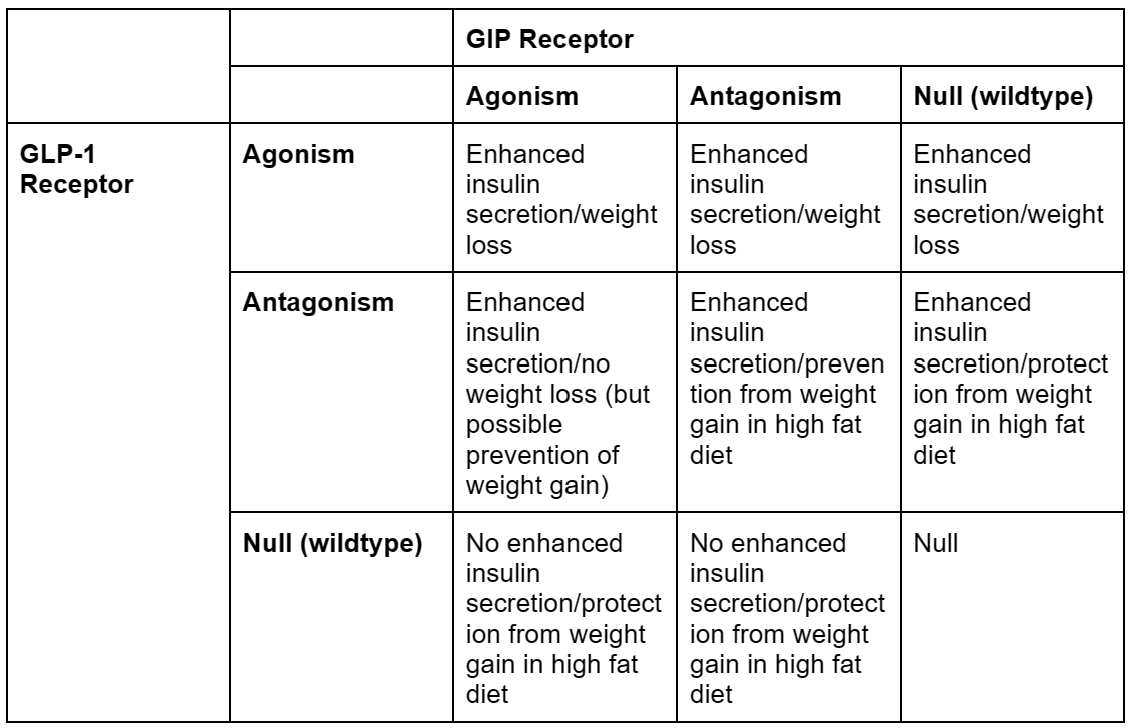
Overall, this seems…very difficult to understand. Now, before I go any further, I did make some simplifications to make this table which may or may not have been justified.
First of all, the distinction between weight loss on a normal diet and protection from weight gain in a high fat diet is a tricky one, because it’s hard to tell if the latter actually qualifies for our final goal, which is presumably “will an obese person taking this lose weight?”. An ideal experiment would likely be to start the mice in any one of the “protection” cells in the table on a high fat diet with no drug, then give them the “protection” drug midway through the trial while keeping them on a high fat diet, and see if they lose weight or maintain their obesity. However, I’m not aware of anyone that’s done that.
If protection from weight gain is removed from the table, the table becomes way more straightforward, with all GLP-1 agonism resulting in weight loss, ignoring the scale (heh) of weight loss, which is a different story.
Second, in this table, I combined gene editing to remove GLP-1 or GIP receptors with pharmacological antagonists for GLP-1 or GIP receptors. This isn’t necessarily the same thing, as genetically deleting receptors is, by definition, permanent and throughout the body, and pharmacological antagonism isn’t1. Pharmacological antagonism is, of course, the only thing we’d be doing in humans, so this may not be justified.
But, if we accept these simplifications, the story we seem to get is that GLP-1 receptor agonism combined with anything results in weight loss. GIP receptor anything (agonism or antagonism) results in protection from weight gain. And GLP-1 anything (agonism or antagonism) results in enhanced insulin secretion.
The review paper I got this data from suggests that the most likely explanation for this is that there is some other system that we aren’t accounting for. So antagonism or genetic deletion take those receptors out of the equation, then whatever other system causes enhanced insulin secretion or protection from weight gain2.
I want to add onto that by noting a couple things.
The first is that insulin’s connection to obesity is complicated, as I’ve talked about before in the context of blind, obese, long-lived cavefish3. Saying that GLP-1 agonists and antagonists cause enhanced insulin secretion is all well and good, but the quick connection of increased insulin to less glucose in the bloodstream to less body fat can’t be made so quickly. Insulin resistance, which is associated with obesity (but quickly goes away in obese people who lose even a tiny amount of weight and then returns when they gain weight again), is not caused by too much insulin, but is likely part of some bodily switch that goes from normal body types to obese body types, like in the cavefish linked above.
This bodily switch is likely, in my opinion, linked to whatever additional system is at play when GLP-1 receptor antagonists cause enhanced insulin secretion and protection from weight gain. In fact, it seems to be the reverse of it: insulin resistance makes it very difficult to lose weight, while GLP-1 receptor antagonism makes it very difficult to gain weight. If I’m right, I would guess that GLP-1 receptor antagonism affects the insulin receptors in cells as well, so that would be the first place I’d look.
The second is that how weight loss happens is, again, complicated. There is definitely some extent to which weight loss happens in the body, which is complicatedly related to insulin and insulin resistance as above. There is also some extent to which weight loss happens in the brain, especially when it comes to GLP-1 agonists and GIP agonists, as mentioned in footnote 1.
And, given that the brain affects weight loss, this means that all of the weird ways that receptors work in the brain still apply. Brain receptors are really weird and can be paradoxical. Take SSRIs (selective serotonin reuptake inhibitors), for instance. They supposedly work by preventing the reuptake of serotonin from the synaptic cleft. This lets them help people with depression and anxiety.
Yet, for some reason, SSRIs usually take a few weeks to really work, even though they start preventing the reuptake of serotonin immediately. Why do they take a few weeks? Nobody knows! It probably has something to do with long term desensitization of the serotonin receptors due to an excess of serotonin, but in order to determine that, we’d have to have a good idea of what serotonin even does, which we don’t.
So, it’s entirely possible GIP receptor agonism acts the same way as GIP receptor antagonism because it’s desensitizing receptors over an extended period of time. That can happen! The only way to check is to dose mice with normal levels of these drugs over an extended period of time, and then check their receptors. Unfortunately, the only studies that have been done to show anything close to this have been with very high doses of these drugs over a limited period of time, which has questionable physiological relevance.
Anyways, unfortunately, all of this is to say that this is all…complicated. I don’t have great answers for what’s going on with GLP-1/GIP agonism/antagonism, only hypotheses. This does make me want to dive in the data and see if I can develop hypotheses regarding the scale and durability of the weight loss after cessation of treatment, because that seems to be the next big battleground, but that’s a story for another blog post.
Adding to this, there are also a number of experiments testing just removing GLP-1 receptors in the pancreas vs. removing them in the whole body to see where weight loss is coming from. Currently, the idea seems to be that weight loss probably comes from acting on the GLP-1 or GIP receptors in the brain (as I discuss in this post), which seems unsurprising to me (people want to eat less) but apparently is a big source of controversy in the field.
I guess they didn’t think semaglutide would affect the brain because it shouldn’t cross the blood-brain barrier, but I’ve thought for a long time that’s an oversimplification. There are a bunch of drugs that affect the brain (like antibiotics or even just straight GABA) that don’t cross the blood-brain barrier, and I don’t buy that it’s because there’s a “second brain in our gut”.
I ignored these complications while making my table as well.
Although “protection from weight gain” seems like a weird way to think about it. Protection is loaded language, because it automatically suggests weight gain is bad, which would definitely not be true from an evolutionary perspective. Maybe something more like “keeps body at a set point” as a control system metaphor would be better.
As a quick refresher, there are normal fish that live in streams outside of caves, and there are weird, obese, long-lived, eyeless cavefish that live in caves. The normal fish have normal metabolisms; the cavefish have weird metabolisms (including insulin resistance) that predispose them to becoming obese. Even though the normal fish and the troglodytic cavefish look incredibly different, they are actually very genetically similar, and normal fish can become cavefish in just a few generations. This suggests that there’s a pretty simple switch somewhere that turns normal fish into obese cavefish.